
Une équipe française évoque l’encéphalopathie mitochondriale
neuro-gastro-intestinale (Mitochondrial Neuro Gastro Intestinal
Encephalomyopathy, MNGIE)[1],
une maladie « extrêmement méconnue des médecins » puisque,
précisent les auteurs, cette affection donne lieu à « une moyenne
de 12 ans d’errance diagnostique et thérapeutique. »
Une triade clinique « évocatrice »
Il s’agit d’une maladie autosomique récessive, liée à la mutation d’un gène (dit TYMP) codant pour une enzyme impliquée dans la régulation des nucléotides pyrimidiques, la thymidine phosphorylase. S’appuyant sur des publications dans les bases de données PubMed et Google Scholar, les auteurs incitent à penser à cette affection « multisystémique largement sous-diagnostiquée » comme une possibilité de diagnostic différentiel lors d’une « anorexie mentale atypique. » Une triade clinique permet de suspecter ce diagnostic difficile où un contexte génétique et enzymatique prend ainsi le masque d’une problématique psychiatrique, l’anorexie mentale dont la dimension somatique retient certes l’attention du médecin à titre de conséquence, mais rarement comme facteur étiologique :
–dysmotilité intestinale (vomissements, nausées, douleurs, voire
« syndrome subocclusif ») ;
–ptosis ou ophtalmoplégie (trouble de la motricité oculaire)
;
–neuropathie périphérique.
Confirmation par les examens complémentaires et surtout le séquençage du gène TYMP
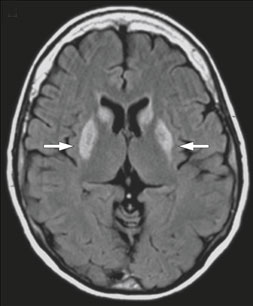
La perte de la fonction thymidine phosphorylase entraînant l’augmentation des taux plasmatiques de thymidine et de déoxyuridine (décelable par des analyses de sang), certains examens complémentaires peuvent « conforter l’hypothèse de MNGIE » (gaz du sang artériel, à la recherche d’une hyperlactacidémie et IRM cérébrale, pour révéler une leucoencéphalopathie « habituellement asymptomatique »), mais le diagnostic est confirmé par d’autres investigations, plus spécifiques :
–constat d’une baisse significative (au moins 10 %) de
l’activité thymidine phosphorylase dans les leucocytes ;
–recherche d’une augmentation de la thymidine (> 3 μmol/L) et de
la déoxyuridine plasmatique (> 5 μmol/L) ;
–et surtout le séquençage par PCR du gène TYMP, pour mettre en
évidence une mutation apportant le diagnostic positif de cette
maladie : on a observé jusqu’à présent « plus de 60
mutations » différentes, chez quelques centaines de patients
concernés.
La perte de la fonction thymidine phosphorylase entraîne aussi une « instabilité (délétions, mutations) de l’ADN mitochondrial » impactant alors « la plupart des organes » où cette activité enzymatique devrait s’exprimer : tube digestif, système nerveux central et périphérique, etc. Cela explique la diversité symptomatologique et la dénomination de cette maladie orpheline qui confirme l’unité de la médecine où la compréhension d’une réalité complexe impose de dépasser la division classique en spécialités étanches.
[1] http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=FR&Expert=298
Dr Alain Cohen
